
You are using an unsupported browser. Please use the latest version of
Chrome, Firefox, Safari or Edge.

Stories
Inspiration. You can find it in every Mass General story. In every hand we hold, and every community partnership and global relief effort we launch. In the face of our newest medical resident and in our latest research breakthrough, decades in the making.
These stories capture how Mass General is harnessing inspiration to make a difference in the lives of patients, families, and our local and global neighbors. Read on. Get inspired. Join us.
Profile in Medicine
In Service to Those Who Served

Hospital News
Recovery Week: Combatting Substance Use Disorder Among Construction Professionals

Patient Story
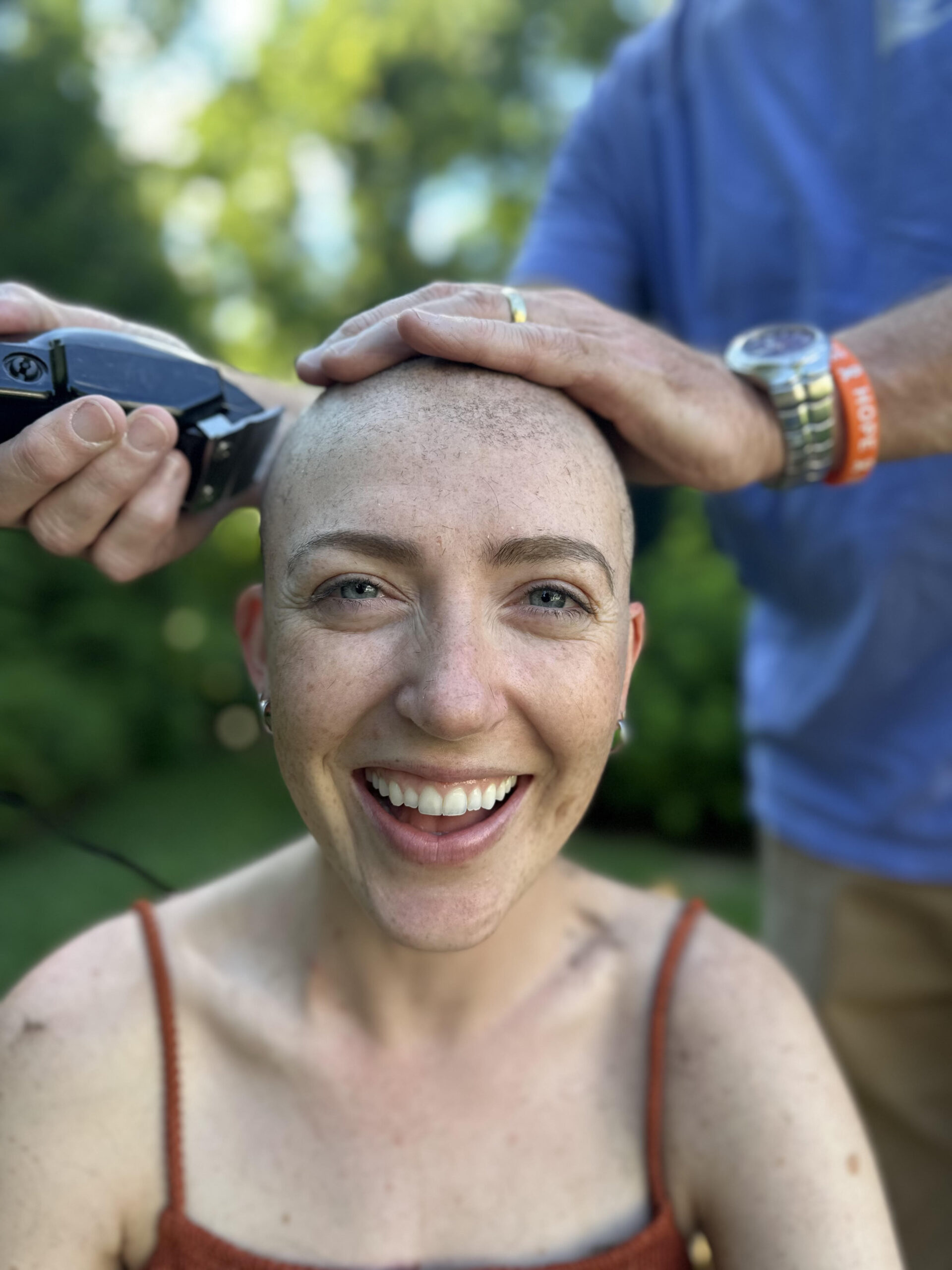
Adolescent and Young Adult Cancer Program: Connecting When it Counts
Hospital News
Xenotransplant Patient Honored at Red Sox Game

Profile in Medicine
Resilience: A Lifelong Journey for Mass General’s New President

Donor Story
Why I Give | Colette Awad

Innovation Story
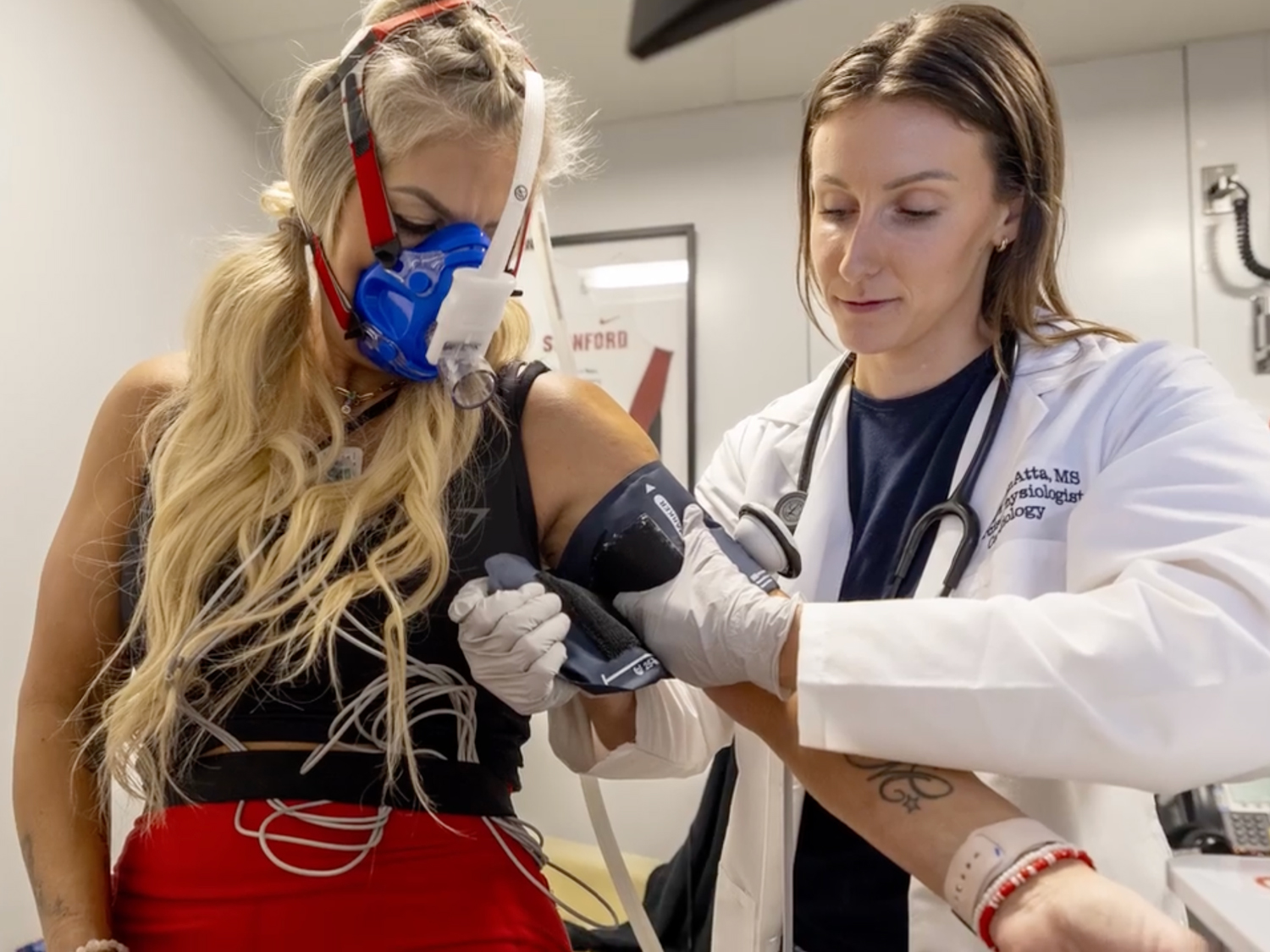
Putting Women’s Heart Health Front and Center
Expert Advice
The Safest Ways to Enjoy Sunny Days

Email Capture
Donor Story
